A PROGRESSIVE AND FATAL DISEASE
Duchenne muscular dystrophy (DMD) affects about 300,000 boys worldwide, with approximately 20,000 cases in the United States.
Most people live out their entire lives without ever hearing the words “Duchenne muscular dystrophy,” with good reason. Most of us will live out our lives without ever knowing a family that has lost – or will lose - a child to Duchenne. We may never hear of a friend or a casual acquaintance who lost a child to Duchenne. Most of us will live out our lives without ever seeing a child with Duchenne.
And there’s a good reason for that, too.
Duchenne muscular dystrophy is one of the rarest diseases on Earth. Children are born with it - only one in every 3,500 live births (mostly in boys). In the United States, with a population of over 330,000,000 people, only 20,000 children are stricken with Duchenne. There is no cure.
This is not the usual type of muscular dystrophy about which we do hear on TV telethons, etc. Most patients with muscular dystrophy manage to live until at least middle-age, and some even make it to old age.
Duchenne muscular dystrophy is different. Typically, boys with Duchenne lose their ability to walk between the ages of ten and fourteen. By their late teens, they lose the strength in their upper bodies, including the ability to move their arms. The disease also affects the heart and breathing muscles, so around this time they also usually need help with breathing at night. Over time, their respiratory systems weaken, and they require constant support. Young men with Duchenne do have a shorter life expectancy but advances in the management of the condition have increased life span significantly and enabled young men to lead much more independent lives than previously possible.
Exon Skipping for DMD
In Duchenne muscular dystrophy, the child’s exons (sections of DNA that code for the protein dystrophin) are deleted, duplicated, or cause an abrupt stop in the protein production process. The impaired exons then interfere with the rest of the gene being pieced together, which results in a mutation that causes the body to lack the protein needed to protect and rebuild muscle fibers. This leads to continuous muscle degeneration and premature death.
What is Exon Skipping?
Genes are built of exons – pieces that must match their neighbors like a jigsaw puzzle. When a gene is read, the exons are assembled together in a process called splicing. When an exon is deleted this can stop pieces from joining together and prevent cells from producing dystrophin protein.
Becker muscular dystrophy – a milder condition also caused by mutations in the dystrophin gene – is caused by deletions where the pieces can still be joined. This allows cells to produce a smaller protein that retains some function.
Exon skipping uses small drugs called antisense oligonucleotides to help cells skip over a specific exon during splicing. This allows cells to join a different set of exons together to produce a protein that is shorter than usual but may have some function.
Pictured below, exon 50 has been deleted. By using a drug to skip over exon 51, the cell can join exon 49 and 51 to produce a shorter than normal dystrophin protein.
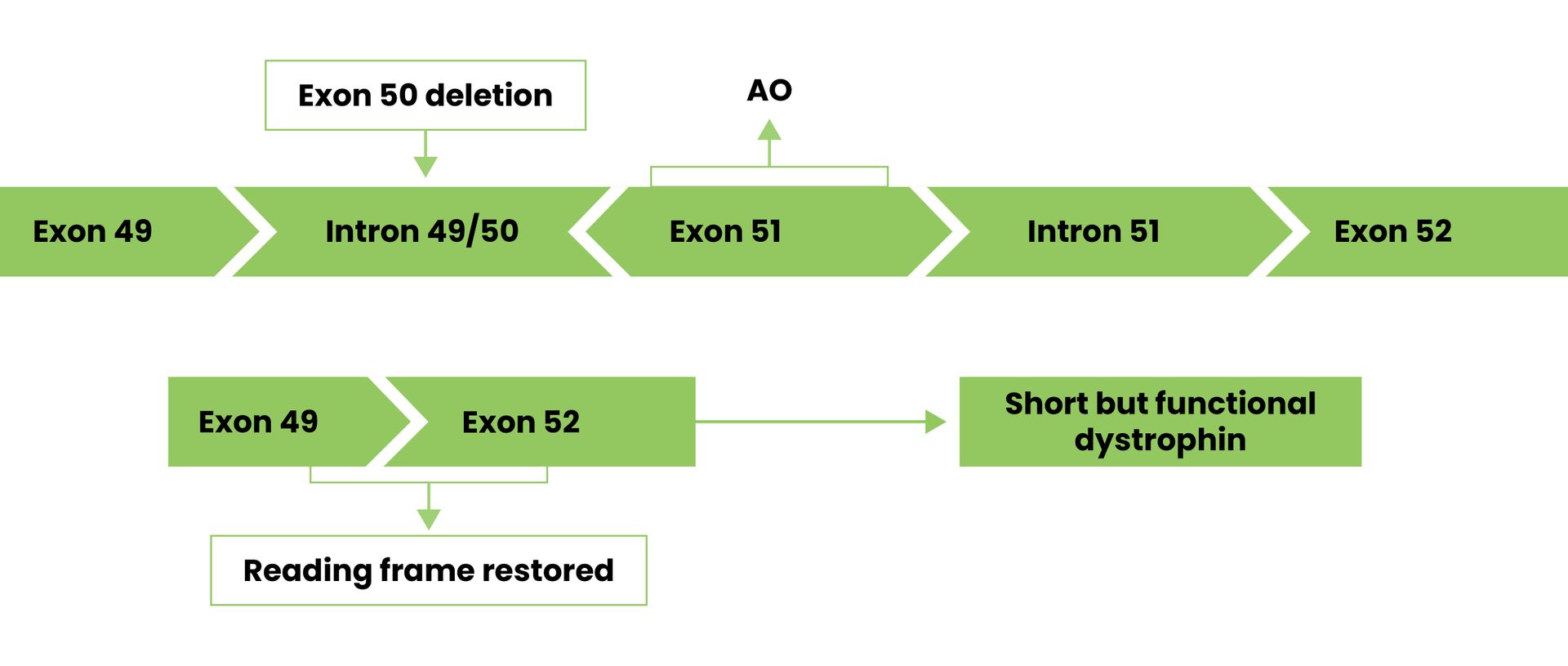
Exon skipping is a potential treatment approach for correcting and restoring production of dystrophin.
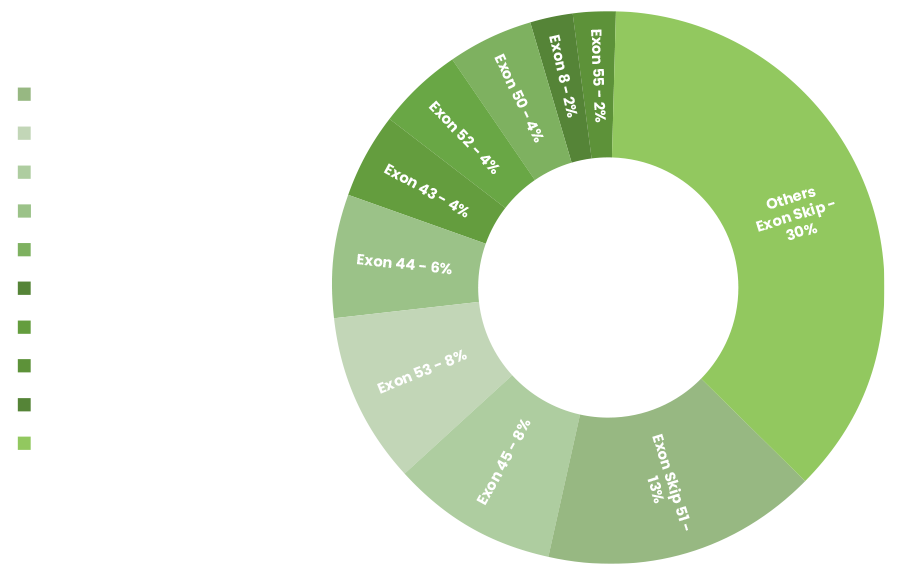
For specific genetic mutations, it allows the body to make a shorter, usable dystrophin. Exon skipping is not a cure for Duchenne, but it may make the effects less severe. No single drug will help everyone with Duchenne. The most common mutation amenable to exon skipping is exon 51, which only affects 13% of all patients. But doctors believe that 60-80% of Duchenne patients may eventually benefit from exon skipping.
Pictured: Duchenne Population Amenable to Exon Skipping. Data suggest up to 80% of patients have genotypes amenable to exon skipping.
Common Questions
See some common questions and answers about Exon Skipping below.
This document contains theoretical and documented, mutations potentially amenable to exon skipping. Not all deletions have been studied and this list may not be complete.
This is an educational resource to provide information about exon skipping only. Please contact your child’s physician or genetic counselor for more information.
Duchenne population amenable to exon skipping was determined through the following source:
- Fletcher, S., et. al. Dystrophin Isoform Induction In Vivo by Antisense-mediated Alternative Splicing. The American Society of Gene & Cell Therapy. 2010;18(6):1218-1223.
- Annemieke Aartsma-Rus, et al. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy. Hum Mutat. 2009 Mar;30 (3):293-9.